文章目录
- salmon转录本定量
- brief
- 模式一:fastq作为输入文件
- 需要特别注意得地方
- 模式二: bam文件作为输入
salmon转录本定量
brief
第一点是,通常说的转录组分析其中有一项是转录本定量,这是一个很trick的说话,说成定量/quantify要适合一些。
因为我们可以根据 reads summary的方式分为两种定量,一种是 transcript-level quantify,一种是 gene-level quantify。
第二点 transcript-level quantify根据比对方式又可以细分:
Transcript quantification 大致分为两类:
- Alignment-based transcript quantification
这里的比对也要作出区分,一种是和基因组比对(aligns reads to the reference genome),一种是和转录组比对(aligns reads to the reference transcriptome)。
和基因组比对后,可以利用Cufflinks or StringTie 等tools,不仅可以测算已知转录本丰度还可以发现新的转录本
和转录组比对后,可以利用RSEM or eXpress or Salmon-Aln 等tools进行转录本丰度的测算 - Alignment-free transcript quantification
就是直接 assign reads directly to transcripts,比如ailfish, Salmon-SMEM,quasi-mapping, and kallisto 这些工具可以实现。
conda activate NGS
conda config --add channels bioconda
conda search salmon
conda install salmon
salmon --help
salmon v0.14.1
Usage: salmon -h|–help or
salmon -v|–version or
salmon -c|–cite or
salmon [–no-version-check] [-h | options]Commands:
index Create a salmon index
quant Quantify a sample
alevin single cell analysis
swim Perform super-secret operation
quantmerge Merge multiple quantifications into a single file
模式一:fastq作为输入文件
# step 1
# make salmon map index
# The index is a structure that salmon uses to quasi-map RNA-seq reads during quantification.
wget https://ftp.ensembl.org/pub/release-111/fasta/macaca_mulatta/cdna/Macaca_mulatta.Mmul_10.cdna.all.fa.gz
# 软件作者希望制作 decoy awere transcriptome,我没管,直接运行下下面的口令
salmon index -t ./Macaca_fascicularis.Macaca_fascicularis_6.0.cdna.all.fa.gz -i ./Macaca_fascicularis.Macaca_fascicularis_6.0.cdna.all.salmon.index
ls Macaca_fascicularis.Macaca_fascicularis_6.0.cdna.all.salmon.index/
# duplicate_clusters.tsv hash.bin header.json indexing.log quasi_index.log refInfo.json rsd.bin sa.bin txpInfo.bin versionInfo.json
# make index done
# step 2
# rawdata QC
datadir="/public/Project_datasets/HY0007/Macaca_fascicularis_RNA-seq/PM-XS05KF2023080038-05四川大学6个食蟹猴普通真核有参转录组建库测序分析任务单/ANNO_XS05KF2023080038_PM-XS05KF2023080038-05_2023-11-02_16-01-18_22C2TNLT3/Rawdata"
mkdir 20240426-NGS-6samples
cd 20240426-NGS-6samples/
mkdir qc
ls ${datadir}/ |while read id ;do echo ${datadir}/${id}/${id}_R1.fq.gz;done > qc.txt
ls ${datadir}/ |while read id ;do echo ${datadir}/${id}/${id}_R2.fq.gz;done >> qc.txt
cat qc.txt |while read id ;do (fastqc -o ./qc $id &);done
multiqc ./qc -o ./multiqc
# trim-adaptor
###
cat qc.txt |grep "R1" >1.txt
cat qc.txt |grep "R2" >2.txt
paste 1.txt 2.txt > trim.txt
cat trim.txt |while read id;do a=($id) && echo /public/home/djs/software/TrimGalore-master/trim_galore -q 25 --phred33 --stringency 3 -o ./Clean_data --paired ${a[0]} ${a[1]}; done > parafly.txt
# conda install -c bioconda parafly
nohup ParaFly -c parafly.txt -CPU 15 >>out.log 2>>err.log &
###
cd Clean_data
mkdir {Fastqc,Multiqc}
ls |grep "val.*fq" |while read id ;do (fastqc -o ./Fastqc ./$id &);done
multiqc ./Fastqc -o ./Multiqc
# step3 map to reference
index="/public/home/djs/reference/macaca_fascicularis/Macaca_fascicularis.Macaca_fascicularis_6.0.cdna.all.salmon.index/"
# 双端测序
cd /public/home/djs/huiyu/project/HY0007/20240426-NGS-6samples/Clean_data
ls *val*gz|cut -d"_" -f 1|sort -u |while read id;do
echo salmon quant -i $index -l ISF --gcBias \
-1 ${id}_R1_val_1.fq.gz -2 ${id}_R2_val_2.fq.gz -p 2 \
-o ../salmon_output/${id}_output
done > paired_salmon.sh
nohup ParaFly -c paired_salmon.sh -CPU 12 >>out.log 2>>err.log &
# quantify result
ls
# aux_info cmd_info.json lib_format_counts.json libParams logs quant.sf
head quant.sf
# ENSMFAT00000064841.2 354 110.000 37.761624 31.000
# ENSMFAT00000064566.2 372 126.000 107.406974 101.000
# ENSMFAT00000061855.2 336 96.000 108.422787 77.680
# ENSMFAT00000061921.2 336 96.000 67.838005 48.603
# ENSMFAT00000061935.2 336 96.000 185.240776 132.717
# ENSMFAT00000097936.1 252 45.460 20.632500 7.000
# ENSMFAT00000064576.2 348 107.929 130.356210 105.000
# ENSMFAT00000064726.2 339 98.000 60.160059 44.000
# ENSMFAT00000064735.2 267 52.000 10.307143 4.000
## 进入R工作
# 进入R做一下 FPKM得转换countToTpm <- function(counts, effLen)
countToTpm <- function(counts, effLen){
rate <- log(counts) - log(effLen)
denom <- log(sum(exp(rate)))
exp(rate - denom + log(1e6))
}
countToFpkm <- function(counts, effLen){
N <- sum(counts)
exp( log(counts) + log(1e9) - log(effLen) - log(N) )
}
fpkmToTpm <- function(fpkm){
exp(log(fpkm) - log(sum(fpkm)) + log(1e6))
}
df <- read.table("quant.sf",header=T)
# 过滤低表达基因
df <- df[df$NumReads >=10,]
# normalization
df$fpkm <- countToFpkm(df$NumReads,df$EffectiveLength)
df$tpm <- countToTpm(df$NumReads,df$EffectiveLength)
write.csv(df,file="quant_filter_transform.csv",row.names=F)
files <- list.files()
dlist <- lapply(files,function(file){read.table(paste("./",file,"/quant_filter_transform.csv",sep=""),header=T,sep=",")})
需要特别注意得地方
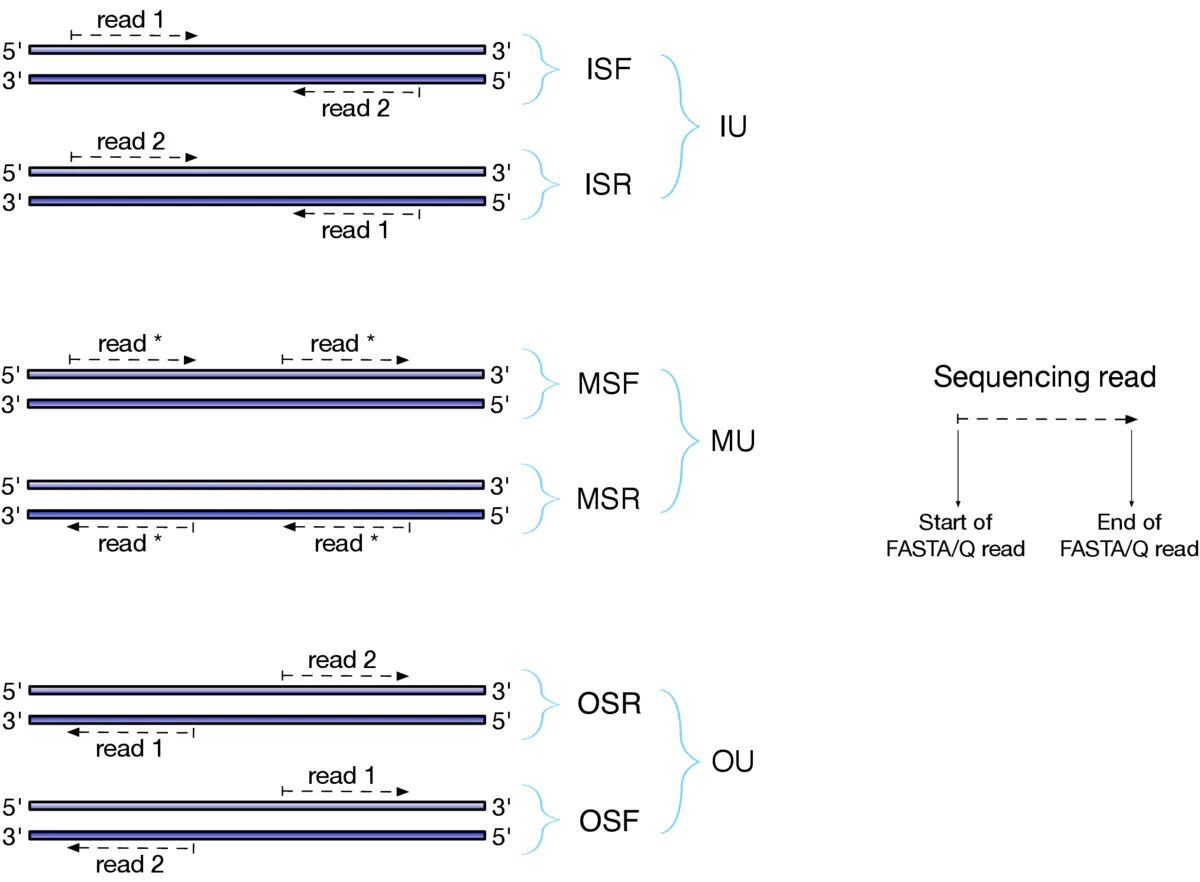
- 参数 —libType A 的设置,一般情况是 --libType ISF 或者 --libType A 让软件自己推测。
模式二: bam文件作为输入
这个bam文件是fastq文件与参考转录组比对的结果,注意不是与参考基因组的比对结果。
然后 transcripts.fa 是参考转录组文件(这种模式下,可以不用建议参考转录组的index)。
> ./bin/salmon quant -t transcripts.fa -l <LIBTYPE> -a aln.bam -o salmon_quant
# quantify result
ls
# aux_info cmd_info.json lib_format_counts.json libParams logs quant.sf
head quant.sf
# ENSMFAT00000064841.2 354 110.000 37.761624 31.000
# ENSMFAT00000064566.2 372 126.000 107.406974 101.000
# ENSMFAT00000061855.2 336 96.000 108.422787 77.680
# ENSMFAT00000061921.2 336 96.000 67.838005 48.603
# ENSMFAT00000061935.2 336 96.000 185.240776 132.717
# ENSMFAT00000097936.1 252 45.460 20.632500 7.000
# ENSMFAT00000064576.2 348 107.929 130.356210 105.000
# ENSMFAT00000064726.2 339 98.000 60.160059 44.000
# ENSMFAT00000064735.2 267 52.000 10.307143 4.000